Aperçu
Le syndrome de Marfan est une maladie héréditaire qui affecte le tissu conjonctif – les fibres qui soutiennent et ancrent vos organes et autres structures dans votre corps. Le syndrome de Marfan affecte le plus souvent le cœur, les yeux, les vaisseaux sanguins et le squelette.
Les personnes atteintes du syndrome de Marfan sont généralement grandes et minces, avec des bras, des jambes, des doigts et des orteils inhabituellement longs. Les dommages causés par le syndrome de Marfan peuvent être légers ou graves. Si l’aorte – le grand vaisseau sanguin qui transporte le sang de votre cœur vers le reste de votre corps – est touchée, l’affection peut mettre votre vie en danger.
Le traitement comprend généralement des médicaments pour maintenir votre tension artérielle basse afin de réduire la tension sur votre aorte. Un suivi régulier pour vérifier la progression des dommages est vital. De nombreuses personnes atteintes du syndrome de Marfan finissent par avoir besoin d’une chirurgie préventive pour réparer l’aorte.
Symptômes

Longueur des doigts dans le syndrome de Marfan
Les personnes atteintes du syndrome de Marfan ont généralement des doigts particulièrement longs. Il est fréquent que leurs pouces dépassent largement le bord de leurs mains lorsqu’ils serrent le poing.

Bras plus longs dans le syndrome de Marfan
Le syndrome de Marfan est une maladie génétique qui entraîne une longueur inhabituelle des bras, des jambes et des doigts. Votre médecin peut vouloir mesurer l’envergure de vos bras s’il pense que vous pourriez être atteint de cette maladie.
Les signes et les symptômes du syndrome de Marfan peuvent varier considérablement, même parmi les membres d’une même famille, car la maladie peut affecter de nombreuses zones différentes du corps. Certaines personnes ne ressentent que des effets légers, mais d’autres développent des complications potentiellement mortelles.
Les caractéristiques du syndrome de Marfan peuvent inclure :
- Carrure grande et élancée
- Bras, jambes et doigts d’une longueur disproportionnée
- Un sternum qui fait saillie vers l’extérieur ou qui s’enfonce vers l’intérieur
- Un palais haut et arqué et des dents serrées
- Souffles cardiaques
- Une myopie extrême
- Une colonne vertébrale anormalement courbée
- Pieds plats
Quand consulter un médecin
Si vous pensez que vous ou votre enfant pouvez être atteint du syndrome de Marfan, parlez-en à votre médecin ou à votre pédiatre. Si votre médecin soupçonne un problème, il vous orientera probablement vers un spécialiste pour une évaluation plus approfondie.
Causes
Le syndrome de Marfan est causé par un défaut du gène qui permet à votre corps de produire une protéine qui contribue à donner au tissu conjonctif son élasticité et sa résistance.
La plupart des personnes atteintes du syndrome de Marfan héritent du gène anormal d’un parent atteint de la maladie. Chaque enfant d’un parent atteint a une chance sur deux d’hériter du gène défectueux. Chez environ 25 % des personnes atteintes du syndrome de Marfan, le gène anormal ne provient d’aucun des parents. Dans ces cas, une nouvelle mutation se développe spontanément.
Facteurs de risque
Le syndrome de Marfan touche indifféremment les hommes et les femmes et se rencontre dans toutes les races et tous les groupes ethniques. Comme il s’agit d’une maladie génétique, le plus grand facteur de risque du syndrome de Marfan est d’avoir un parent atteint de la maladie.
Complications
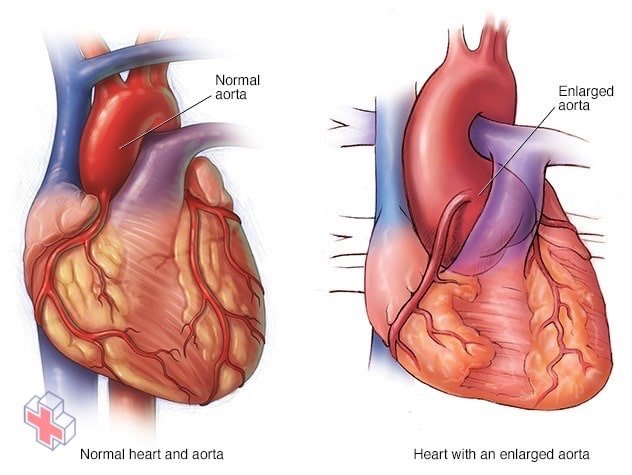
Anévrisme à la racine de l’aorte
La pression du sang quittant votre cœur peut provoquer un renflement de la paroi de votre aorte, comme un point faible dans un pneu. Chez les personnes atteintes du syndrome de Marfan, ce phénomène est le plus susceptible de se produire à la racine de l’aorte, là où l’artère quitte le cœur.
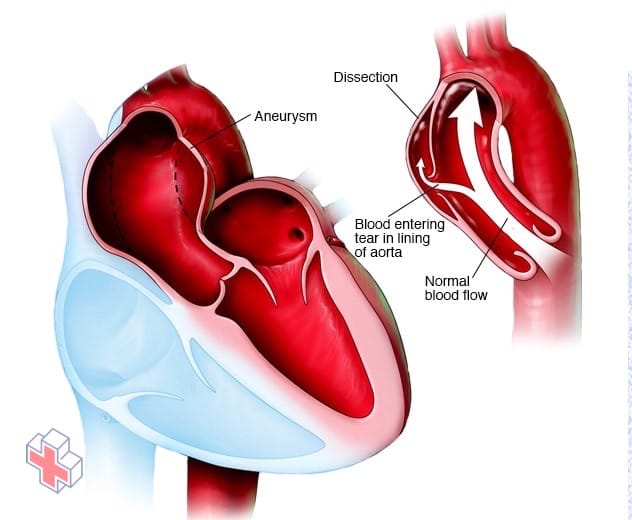
Anévrisme aortique et dissection aortique
Un anévrisme aortique se produit lorsqu’un point faible de la paroi de votre aorte commence à se bomber (à gauche). Cela peut se produire n’importe où dans votre aorte. La présence d’un anévrisme augmente le risque de dissection aortique – une déchirure de la paroi de l’aorte, illustrée sur l’image de droite.
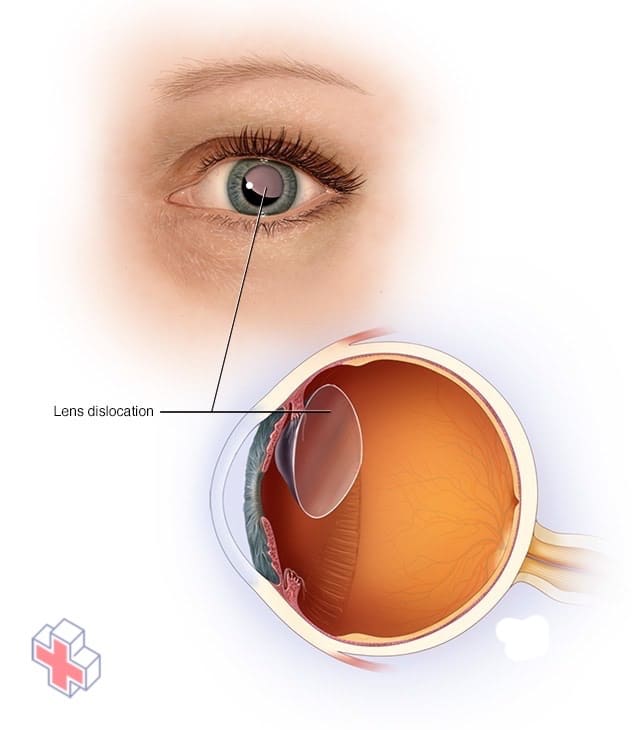
Dislocation de la lentille
Certaines personnes atteintes du syndrome de Marfan peuvent subir une dislocation du cristallin de leur œil.

Détachement de la rétine
Le décollement de la rétine décrit une situation d’urgence dans laquelle une couche critique de tissu (la rétine) à l’arrière de l’œil se détache de la couche de vaisseaux sanguins qui lui fournit l’oxygène et les nutriments. Le décollement de la rétine est souvent accompagné d’éclairs et de flotteurs dans votre vision.
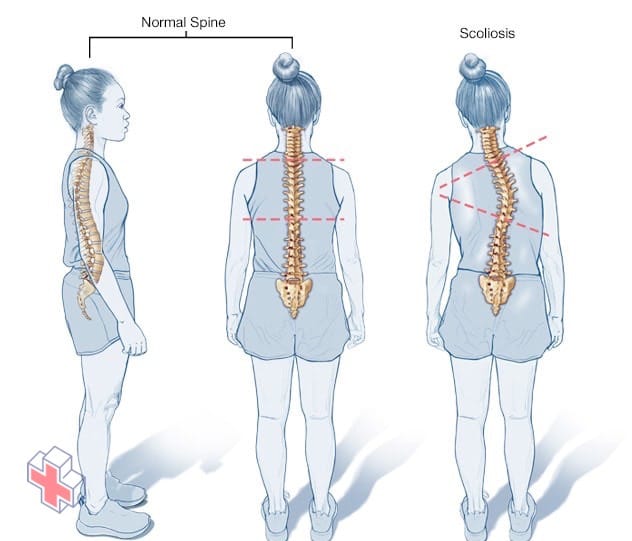
Scoliose
Vue de côté, la colonne vertébrale normale a la forme d’un S allongé, le haut du dos se courbant vers l’extérieur et le bas du dos s’incurvant légèrement vers l’intérieur. En revanche, vue de derrière, la colonne vertébrale doit apparaître comme une ligne droite de la base du cou au coccyx. La scoliose est une courbure latérale de la colonne vertébrale.
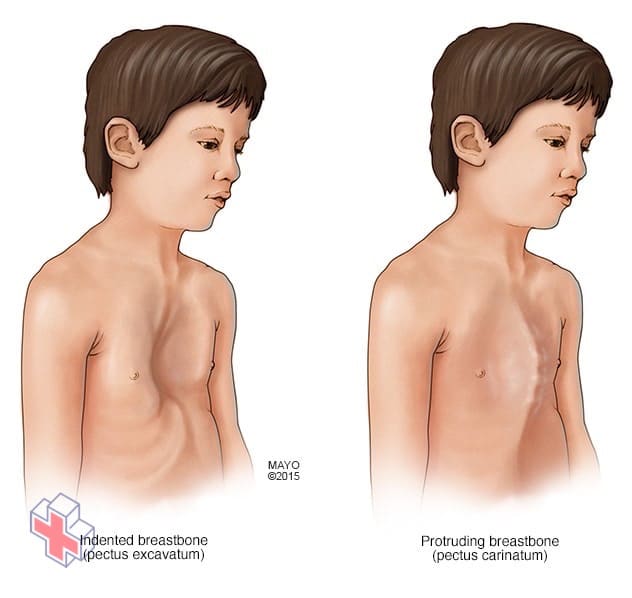
Anomalies thoraciques
Le syndrome de Marfan peut interférer avec le développement normal des côtes, ce qui peut entraîner une saillie du sternum ou une apparence enfoncée dans la poitrine.
Comme le syndrome de Marfan peut affecter presque toutes les parties de votre corps, il peut entraîner une grande variété de complications.
Complications cardiovasculaires
Les complications les plus dangereuses du syndrome de Marfan concernent le cœur et les vaisseaux sanguins. Le tissu conjonctif défectueux peut affaiblir l’aorte – la grande artère qui naît du cœur et alimente le corps en sang.
- Anévrisme de l’aorte. La pression du sang quittant votre cœur peut provoquer un renflement de la paroi de votre aorte, comme un point faible dans un pneu. Chez les personnes atteintes du syndrome de Marfan, ce phénomène est le plus susceptible de se produire au niveau de la racine de l’aorte, là où l’artère quitte le cœur.
- Dissection aortique. La paroi de l’aorte est constituée de couches. La dissection se produit lorsqu’une petite déchirure dans la couche la plus interne de la paroi de l’aorte permet au sang de se presser entre les couches interne et externe de la paroi. Cela peut provoquer une douleur intense dans la poitrine ou le dos. Une dissection aortique affaiblit la structure du vaisseau et peut entraîner une rupture, qui peut être fatale.
- Malformations valvulaires. Les personnes atteintes du syndrome de Marfan peuvent avoir des tissus faibles dans leurs valves cardiaques. Cela peut entraîner un étirement du tissu valvulaire et un fonctionnement anormal des valves. Lorsque les valves cardiaques ne fonctionnent pas correctement, votre cœur doit souvent travailler plus fort pour compenser. Cela peut éventuellement conduire à une insuffisance cardiaque.
Complications oculaires
Les complications oculaires peuvent inclure :
- Dislocation du cristallin. La lentille de focalisation de votre œil peut se déplacer si ses structures de soutien s’affaiblissent. Le terme médical pour ce problème est ectopia lentis, et il se produit chez plus de la moitié des personnes atteintes du syndrome de Marfan.
- Problèmes rétiniens. Le syndrome de Marfan augmente également le risque de décollement ou de déchirure de la rétine, le tissu sensible à la lumière qui tapisse la paroi arrière de votre œil.
- Glaucome ou cataracte à apparition précoce. Les personnes atteintes du syndrome de Marfan ont tendance à développer ces problèmes oculaires à un plus jeune âge. Le glaucome entraîne une augmentation de la pression à l’intérieur de l’œil, ce qui peut endommager le nerf optique. Les cataractes sont des zones nuageuses dans le cristallin normalement clair de l’œil.
Complications squelettiques
Le syndrome de Marfan augmente le risque de courbures anormales de la colonne vertébrale, comme la scoliose. Il peut également interférer avec le développement normal des côtes, ce qui peut entraîner une saillie du sternum ou une apparence enfoncée dans la poitrine. Les douleurs aux pieds et les lombalgies sont fréquentes avec le syndrome de Marfan.
Complications de la grossesse
Le syndrome de Marfan peut affaiblir les parois de l’aorte, l’artère principale qui quitte le cœur. Pendant la grossesse, le cœur pompe plus de sang que d’habitude. Cela peut exercer un stress supplémentaire sur l’aorte, ce qui augmente le risque de dissection ou de rupture mortelle.
Diagnostic
Le syndrome de Marfan peut être difficile à diagnostiquer pour les médecins car de nombreux troubles du tissu conjonctif présentent des signes et des symptômes similaires. Même parmi les membres d’une même famille, les signes et symptômes du syndrome de Marfan varient considérablement – tant dans leurs caractéristiques que dans leur gravité.
Certaines combinaisons de symptômes et d’antécédents familiaux doivent être présentes pour confirmer un diagnostic de syndrome de Marfan. Dans certains cas, une personne peut présenter certaines caractéristiques du syndrome de Marfan, mais pas suffisamment pour que le diagnostic soit posé.
Tests cardiaques
Si votre médecin soupçonne un syndrome de Marfan, l’un des premiers tests qu’il peut recommander est une échocardiographie. Ce test utilise des ondes sonores pour capturer des images en temps réel de votre cœur en mouvement. Il permet de vérifier l’état de vos valves cardiaques et la taille de votre aorte. Les autres options d’imagerie cardiaque comprennent la tomographie par ordinateur (CT) et l’imagerie par résonance magnétique (IRM).
Si vous êtes diagnostiqué avec le syndrome de Marfan, vous devrez passer régulièrement des examens d’imagerie pour surveiller la taille et l’état de votre aorte.
Examens des yeux
Les examens oculaires qui peuvent être nécessaires comprennent :
- Examen à la lampe à fente. Ce test vérifie la présence d’une dislocation du cristallin, de cataractes ou d’une rétine décollée. Vos yeux devront être complètement dilatés à l’aide de gouttes pour cet examen.
- Test de pression oculaire. Pour vérifier l’absence de glaucome, votre ophtalmologue peut mesurer la pression à l’intérieur de votre globe oculaire en le touchant avec un outil spécial. Des gouttes ophtalmiques anesthésiantes sont généralement utilisées avant ce test.
Tests génétiques
Les tests génétiques sont souvent utilisés pour confirmer le diagnostic du syndrome de Marfan. Si une mutation Marfan est trouvée, les membres de la famille peuvent être testés pour voir s’ils sont également affectés. Vous voudrez peut-être parler à un conseiller en génétique avant de fonder une famille, pour voir quelles sont vos chances de transmettre le syndrome de Marfan à vos futurs enfants.
Traitement
Bien qu’il n’existe pas de remède pour le syndrome de Marfan, le traitement se concentre sur la prévention des diverses complications de la maladie. Pour ce faire, vous devrez être contrôlé régulièrement pour détecter les signes de progression des dommages causés par la maladie.
Par le passé, les personnes atteintes du syndrome de Marfan mouraient souvent jeunes. Grâce à un suivi régulier et à un traitement moderne, la plupart des personnes atteintes du syndrome de Marfan peuvent désormais espérer vivre une durée de vie plus normale.
Médicaments
Les médecins prescrivent souvent des médicaments hypotenseurs pour empêcher l’aorte de s’élargir et pour réduire le risque de dissection et de rupture.
Thérapie
Les problèmes de vision associés à un cristallin disloqué dans l’œil peuvent souvent être corrigés par des lunettes ou des lentilles de contact.
Interventions chirurgicales et autres

Procédure d’anévrisme de la racine de l’aorte ascendante
Une procédure d’anévrisme de la racine aortique ascendante peut être effectuée de deux manières. Dans le cas d’un remplacement de la racine aortique, votre chirurgien retire une section de votre aorte et votre valve aortique. La section aortique est remplacée par un tube artificiel (greffe). La valve aortique est remplacée par une valve mécanique ou biologique (image en bas à droite). Certaines personnes subissent une réparation de la racine aortique épargnant la valve (image en haut à droite), dans laquelle la valve aortique reste en place.
En fonction de vos signes et symptômes, les procédures peuvent inclure :
- Réparation de l’aorte. Si le diamètre de votre aorte atteint environ 50 millimètres (2 pouces) ou si elle s’élargit rapidement, votre médecin peut recommander une opération visant à remplacer une partie de votre aorte par un tube en matériau synthétique. Cela peut aider à prévenir une rupture potentiellement mortelle. Il se peut que votre valve aortique doive également être remplacée.
- Traitement de la scoliose. En cas de scoliose importante, une consultation avec un spécialiste de la colonne vertébrale est nécessaire. L’attelle et la chirurgie sont nécessaires dans certains cas.
- Corrections du sternum. Des options chirurgicales sont disponibles pour corriger l’apparence d’un sternum enfoncé ou saillant. Comme ces opérations sont souvent considérées comme étant à des fins esthétiques, il se peut que votre assurance ne couvre pas les frais.
- Chirurgie des yeux. Si des parties de votre rétine se sont déchirées ou détachées de l’arrière de votre œil, une réparation chirurgicale est généralement réussie. Si vous avez des cataractes, votre cristallin opacifié peut être remplacé par une lentille artificielle.
Mode de vie et remèdes maison
Vous devrez peut-être éviter les sports de compétition et certaines activités récréatives si vous présentez un risque accru de dissection ou de rupture aortique. L’augmentation de la pression sanguine, fréquente dans des activités telles que l’haltérophilie, exerce une pression supplémentaire sur l’aorte. Les activités moins intenses – comme la marche rapide, le bowling, le tennis en double ou le golf – sont généralement plus sûres.
Adaptation et soutien
Vivre avec une maladie génétique peut être extrêmement difficile, tant pour les adultes que pour les enfants. Les adultes peuvent se demander comment la maladie affectera leur carrière, leurs relations et leur perception d’eux-mêmes. Et ils peuvent s’inquiéter de transmettre le gène défectueux à leurs enfants.
Mais le syndrome de Marfan peut être encore plus difficile pour les jeunes, notamment parce que la conscience de soi souvent inhérente à l’enfance et à l’adolescence peut être exacerbée par l’effet de la maladie sur l’apparence, les résultats scolaires et les capacités motrices.
Aider les enfants à faire face
En travaillant ensemble, les parents, les enseignants et les professionnels de la santé peuvent apporter aux enfants un soutien émotionnel et des solutions pratiques pour certains des aspects les plus pénibles de la maladie. Par exemple, les enfants atteints du syndrome de Marfan peuvent avoir des difficultés à l’école en raison de problèmes de vision qui peuvent être corrigés par des lunettes ou des lentilles de contact.
Pour la plupart des jeunes, les préoccupations esthétiques sont au moins aussi importantes que les préoccupations scolaires. Les parents peuvent aider en anticipant ces préoccupations et en proposant des solutions, par exemple :
- Des lentilles de contact au lieu de lunettes
- Un appareil orthopédique pour la scoliose
- Des travaux dentaires pour des dents encombrées
- Des vêtements qui flattent un cadre grand et mince
Groupes de soutien
Les personnes atteintes du syndrome de Marfan trouvent souvent utile de parler avec d’autres personnes confrontées aux mêmes défis. La Fondation Marfan offre une variété de services de soutien en ligne.
Préparation de votre rendez-vous
Le syndrome de Marfan peut affecter de nombreuses parties de votre corps. Il se peut donc que vous deviez consulter divers spécialistes médicaux, tels que :
- Un cardiologue, un médecin spécialisé dans les troubles du cœur et des vaisseaux sanguins
- Un ophtalmologiste, un médecin spécialisé dans les troubles oculaires
- Un orthopédiste, un médecin spécialisé dans les problèmes structurels du squelette
- Un généticien, un médecin spécialisé dans les troubles génétiques
Pour utiliser au mieux le temps du rendez-vous, planifiez à l’avance et ayez à portée de main les informations importantes, notamment :
- Des descriptions détaillées de tous vos symptômes
- Détails de vos antécédents médicaux, y compris toute opération chirurgicale antérieure
- Les rapports de radiographies et d’échocardiogrammes antérieurs, qui peuvent souvent être envoyés par voie électronique
- Une liste de tous vos médicaments et suppléments
Ce que vous pouvez attendre de votre médecin
Tous vos médecins voudront connaître vos symptômes spécifiques et savoir si quelqu’un dans votre famille a été atteint du syndrome de Marfan ou a connu une invalidité ou un décès précoce et inexpliqué lié au cœur.
 »
»
Laisser un commentaire